SMA Tedavisinde On Yıl: Genetik Devrimden Uzun Vadeli Bakım Sorularına
Spinal musküler atrofi (SMA), on yıl önce klinik pratiğin en ağır nöromüsküler hastalıklarından biri olarak görülüyordu; çoğu vakada tablo, hızlı motor nöron kaybı, ilerleyici kas güçsüzlüğü ve giderek artan bakım gereksinimiyle tanımlanıyordu. Ancak son 10 yılda bu manzara kökten değişti. SMN1 genindeki delesyonlar veya mutasyonlarla ilişkili olan bu otozomal resesif hastalıkta, SMN proteini eksikliğinin moleküler temeline doğrudan yönelen hastalık düzenleyici tedaviler, SMA’nın doğal seyrini tarihte ilk kez anlamlı biçimde dönüştürdü. Bugün uzmanlar, bu alanı yalnızca tedavi başarısı olarak değil, aynı zamanda genetik hastalıkların biyolojisinin klinik sonuçlara nasıl çevrilebileceğini gösteren örnek bir dönüşüm olarak değerlendiriyor.
Dönüm noktası, 2016’da ilk tedavinin onaylanmasıyla geldi. Bu gelişme, yalnızca bir ilaç onayı anlamına gelmiyordu; aynı zamanda SMA için “kaçınılmaz ilerleme” döneminin sona erdiğine dair güçlü bir işaretti. Takip eden yıllarda biriken klinik ve gerçek yaşam verileri, bu tedavi yaklaşımlarının güvenlik ve etkinliğine ilişkin kanıt tabanını genişletti. Özellikle en ağır klinik tablolar arasında yer alan tip 1 SMA’da bile, hastalığın doğal seyrine kıyasla belirgin iyileşmeler bildirildi. Yine de uzmanlar, bu ilerlemenin hastalığı tamamen ortadan kaldırdığı şeklinde yorumlanmaması gerektiğini vurguluyor; çünkü uzun dönem sonuçlar, tedaviye başlama zamanı, izlem stratejileri ve farklı fenotiplerde yanıt değişkenliği gibi kritik sorular hâlâ masada duruyor.
SMA’nın biyolojik temeli, SMN1 genindeki bozuklukların motor nöronlar üzerinde yarattığı yıkıcı etkiyle açıklanıyor. Motor nöronların kaybı, istemli kaslarda atrofiye ve ciddi güçsüzlüğe yol açıyor. Bu mekanizmanın anlaşılması, tedavi geliştirme sürecini de doğrudan şekillendirdi. Son on yılda klinik uygulamaya giren başlıca üç yaklaşım, SMN proteini düzeyini artırmayı hedefleyen farklı stratejiler üzerine kuruldu. İlk grupta, antisens oligonükleotidler yer aldı; bu sınıfta nusinersen, hastalığın moleküler düzenlenmesinde çığır açan bir seçenek olarak öne çıktı. Diğer bir yaklaşım, SMN1 gen kusurunu telafi etmeyi amaçlayan gen temelli tedaviler oldu. Buna ek olarak, SMN protein üretimini artırmaya yönelik ağızdan kullanılan küçük molekül tedaviler de yönetim seçeneklerini genişletti.
Bu tedavi çeşitliliği, SMA bakımını yalnızca “ilaç verme” pratiğinden çıkarıp daha karmaşık bir klinik karar alanına dönüştürdü. Hekimler artık tedavi seçerken hastanın yaşını, hastalık alt tipini, semptomların başlangıç zamanını, motor fonksiyon düzeyini ve izlemde beklenen yükü birlikte değerlendirmek zorunda. Özellikle erken tanı ve mümkünse tarama programlarıyla çok erken dönemde başlanan tedavilerin, klinik sonuçları daha iyi etkileyebileceğine dair genel eğilim, SMA alanında son yılların en önemli pratik derslerinden biri oldu. Bununla birlikte, tüm hastalar aynı ölçüde yanıt vermiyor; bu durum, tedavinin zamanlaması ve kişiselleştirilmiş bakımın önemini artırıyor.
Güncel literatür, hastalığın tedavi edilebilir bir genetik bozukluğa dönüşmesinin yeni sorumluluklar da yarattığını gösteriyor. Uzun süreli güvenlik, tedaviye kalıcı erişim, motor gelişimin sürdürülebilirliği ve multidisipliner destek ihtiyacı, artık SMA bakımının merkezinde yer alıyor. Solunum desteği, beslenme yönetimi, ortopedik izlem ve fizik tedavi gibi destekleyici müdahaleler hâlâ büyük önem taşıyor; çünkü hastalık modifiye edici tedaviler tek başına tüm komplikasyonları ortadan kaldırmıyor. Özellikle daha ağır fenotiplerde, nörolojik kazanımlar ile günlük bakım gereksinimleri arasındaki denge, klinisyenlerin dikkatle takip ettiği bir alan olmaya devam ediyor.
Uzmanlar, son on yıldaki ilerlemeyi “bench’ten yatağa” uzanan bir başarı öyküsü olarak tanımlasa da, bu öykünün finali henüz yazılmış değil. SMN1 eksikliğini hedefleyen tedavilerin sağladığı kazanımlar, SMA araştırmalarında yeni sorular doğurdu: Hangi hastalar hangi tedaviden en fazla yarar görüyor? Tedavi ne kadar erken başlanmalı? Uzun vadede etkinlik nasıl korunmalı? Tedavi yanıtı biyobelirteçlerle öngörülebilir mi? Bu soruların her biri, önümüzdeki dönemde hem klinik kılavuzları hem de günlük hasta yönetimini şekillendirecek nitelikte.
Bugün gelinen noktada SMA, artık yalnızca progresif bir kas zayıflığı hastalığı olarak değil, moleküler mekanizmalar hedef alınarak seyrinin değiştirilebildiği bir genetik hastalık olarak anılıyor. 2016’da açılan kapı, on yıl içinde yalnızca yeni ilaçların değil, yeni bir klinik yaklaşımın da önünü açtı. Bu yaklaşım, erken tanı, bireyselleştirilmiş tedavi seçimi ve uzun dönem izlem ihtiyacını aynı çerçevede topluyor. SMA tedavisinde yaşanan dönüşüm, nadir hastalıklar alanında bilimsel bilginin klinik yarara nasıl çevrilebileceğini gösterirken, aynı zamanda kalıcı bakım gereksinimlerinin ve bilinmezliklerin hâlâ devam ettiğini de hatırlatıyor.
 Lipid Bazlı Yardımcı Maddeler Ağızdan İlaç Emilimini Yeniden Şekillendiriyor
Lipid Bazlı Yardımcı Maddeler Ağızdan İlaç Emilimini Yeniden Şekillendiriyor
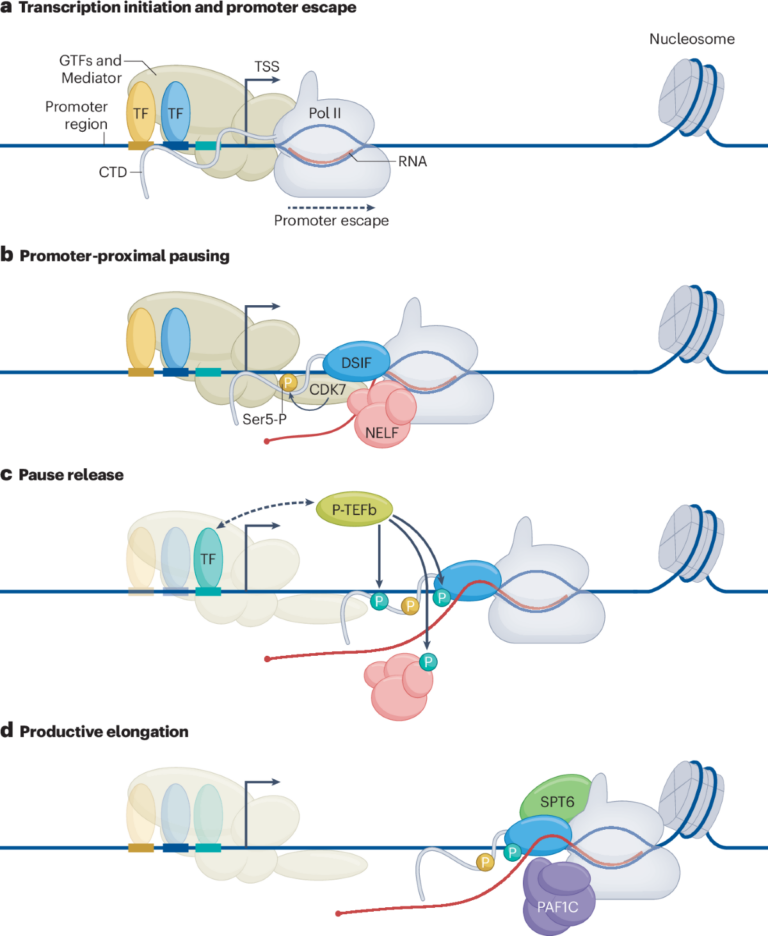 RNA Polimeraz II’nin Gen Okuma Süreci Hastalık Araştırmalarına Yeni Bir Pencere Açıyor
RNA Polimeraz II’nin Gen Okuma Süreci Hastalık Araştırmalarına Yeni Bir Pencere Açıyor
 Kolorektal kanserde ameliyat sonrası riskleri yaş ve tümör evresi birlikte şekillendiriyor
Kolorektal kanserde ameliyat sonrası riskleri yaş ve tümör evresi birlikte şekillendiriyor