Huntington Hastalığında DNA’daki Çift Zincir Kırıkları Hastalığın Yıkıcı Etkisini Körüklüyor
Huntington hastalığının nasıl ilerlediğine dair yerleşik görüşler, son yıllarda genetik düzeydeki yeni bulgularla yeniden şekilleniyor. Nature Communications’ta yayımlanan yeni bir çalışma, hastalığın sinir hücrelerine verdiği zararda DNA’daki çift zincir kırıklarının sanıldığından çok daha merkezi bir rol oynadığını gösterdi. Polyzos, Cheong, Yoo ve çalışma arkadaşlarının yürüttüğü araştırma, mutant huntingtin geninde görülen somatik genişlemenin tek başına açıklayıcı olmadığını; DNA hasarının, genişleme olup olmadığına bakılmaksızın nöronal toksisiteyi tetikleyebildiğini ortaya koydu.
Huntington hastalığı, HTT genindeki CAG tekrar dizisinin uzamasıyla ortaya çıkan kalıtsal bir nörodejeneratif bozukluk olarak biliniyor. Uzamış CAG dizisi, protein içinde anormal derecede uzun bir poliglutamin zinciri oluşmasına yol açıyor ve bu durum özellikle hareket, biliş ve davranışla ilişkili beyin bölgelerinde ilerleyici hasara neden oluyor. Uzun süredir araştırmacılar, hastalık dokularında zaman içinde CAG tekrarlarının daha da artması anlamına gelen somatik genişlemeyi, kötüleşen tabloyu açıklayan başlıca süreçlerden biri olarak değerlendiriyordu. Ancak yeni bulgular, bu çerçevenin tek başına yeterli olmadığını düşündürüyor.
Araştırma ekibi, Huntington hastalığının iyi tanımlanmış bir fare modelinde genetik ve moleküler araçları kullanarak DNA onarımı, somatik genişleme ve nöronal stres arasındaki ilişkileri ayırmaya çalıştı. Elde edilen sonuçlar, çift zincir kırıklarının, mutant huntingtin taşıyan nöronlarda toksisiteyi başlatmak veya hızlandırmak için yeterli olabildiğini gösterdi. Bu etki, somatik genişleme süreçlerinin aktif olup olmadığına bağlı görünmüyordu. Başka bir deyişle, DNA’nın iki ipliğinin birden kırılması, hastalık biyolojisinde sadece eşlik eden bir hasar değil, doğrudan zararlı bir itici güç olabilir.
Çift zincir kırıkları hücreler için son derece kritik hasarlardır. Tek iplikli hasarlar çoğu zaman daha kolay onarılabilirken, çift zincir kırıkları genom bütünlüğünü tehdit eder ve hücreyi çok daha ağır bir stres yanıtına zorlar. Sinir hücreleri ise bölünmeyen, uzun ömürlü ve yüksek enerji ihtiyacı olan hücreler oldukları için bu tür hasarlara özellikle hassastır. Beyin dokusunda DNA onarım süreçlerinin aksaması, yalnızca mutasyon birikimine değil, aynı zamanda hücresel işlevlerin bozulmasına ve nihayetinde hücre ölümüne de katkıda bulunabilir. Çalışmanın en dikkat çekici yönlerinden biri, Huntington hastalığında daha önce ağırlık verilen tekrar genişlemesi modelinin yanına DNA hasarı ve onarım mekanizmalarını güçlü bir şekilde yerleştirmesi oldu.
Yazarlar, somatik genişlemeyi etkileyen etmenleri değiştirerek ve DNA onarım yollarıyla ilişkili mutasyonları kullanarak, toksisitenin hangi koşullarda arttığını ayrıntılı biçimde inceledi. Bu yaklaşım, mutant huntingtin’in neden olduğu zararın yalnızca tekrar dizisinin büyümesiyle açıklanamayacağını, hücrenin DNA hasarına verdiği cevabın da belirleyici olduğunu ortaya koydu. Bulgular, DNA çift zincir kırıklarının birikmesinin nöronal işlev kaybını kolaylaştırabileceğine ve hastalığın moleküler temelinde beklenenden daha erken bir aşamada devreye girebileceğine işaret ediyor.
Bu sonuçlar, Huntington hastalığında tedavi stratejilerinin de yeniden düşünülmesini gerektirebilir. Bugüne kadar geliştirilen birçok yaklaşım, mutant huntingtin proteininin üretimini azaltmaya, CAG tekrarlarının davranışını sınırlamaya veya genetik düzeltmeye odaklandı. Yeni çalışma ise DNA hasar yanıtı yollarının da potansiyel bir müdahale alanı olabileceğini gösteriyor. Elbette bu, doğrudan klinik bir tedavi seçeneğinin ortaya çıktığı anlamına gelmiyor. Ancak bulgular, nöronlarda DNA onarımını düzenleyen mekanizmaların hastalık sürecinin önemli bir parçası olabileceğini ve gelecekte ilaç geliştirme çalışmalarında dikkate alınması gerektiğini düşündürüyor.
Huntington hastalığında DNA hasarının rolü, nörodejenerasyon araştırmalarında daha geniş bir eğilimi de yansıtıyor. Alzheimer ve Parkinson gibi hastalıklarda da DNA onarımı, genomik stabilite ve hücresel stres yanıtlarının sinir hücresi kaybıyla ilişkili olabileceğine dair kanıtlar artıyor. Bu yeni çalışma, Huntington hastalığında da benzer bir biyolojik eksenin bulunduğunu ve genom hasarının yalnızca sonuç değil, hastalık sürücüsü olabileceğini destekliyor. Özellikle uzun ömürlü nöronlarda biriken çift zincir kırıkları, hastalığın ilerleyici doğasını açıklamada önemli bir parça sunuyor.
Yine de araştırmanın klinik yorumunda dikkatli olmak gerekiyor. Bulgular, bir fare modelinden elde edildi ve insan hastalığındaki süreçlerin tüm ayrıntıları birebir aynı olmak zorunda değil. Buna rağmen çalışma, hastalığın moleküler patogenezine dair uzun süredir süren tartışmada güçlü bir veri sunuyor. Somatik genişleme ile DNA hasarı arasındaki ilişkinin yeniden tanımlanması, Huntington hastalığının neden bazı hücre tiplerinde daha ağır seyrettiğini anlamaya da yardımcı olabilir. Bu da ileride daha seçici, mekanizmaya dayalı tedavilerin geliştirilmesini kolaylaştırabilir.
Sonuç olarak, bu bulgular Huntington hastalığı alanında önemli bir bakış açısı değişimine işaret ediyor. DNA çift zincir kırıkları, mutant huntingtin kaynaklı toksisitenin pasif bir eşlikçisi değil; sinir hücrelerinin hayatta kalmasını tehdit eden temel unsurlardan biri olabilir. Araştırma, hastalığın yalnızca tekrar DNA’sının genişlemesi üzerinden değil, aynı zamanda DNA onarım sistemlerinin kırılganlığı üzerinden de anlaşılması gerektiğini ortaya koyuyor.
 Lipid Bazlı Yardımcı Maddeler Ağızdan İlaç Emilimini Yeniden Şekillendiriyor
Lipid Bazlı Yardımcı Maddeler Ağızdan İlaç Emilimini Yeniden Şekillendiriyor
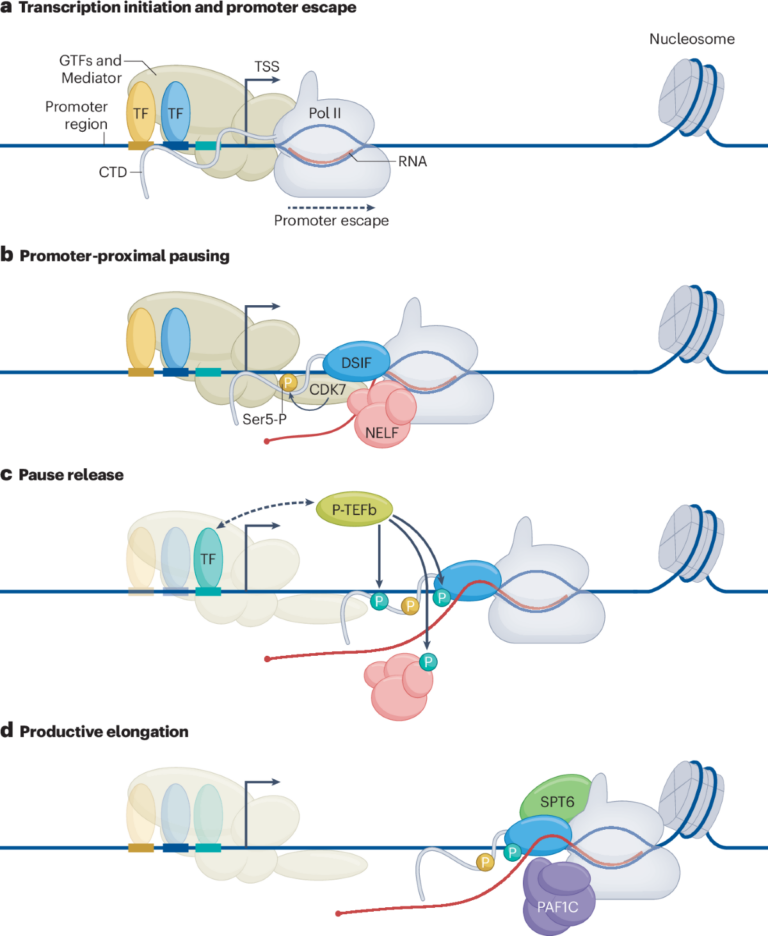 RNA Polimeraz II’nin Gen Okuma Süreci Hastalık Araştırmalarına Yeni Bir Pencere Açıyor
RNA Polimeraz II’nin Gen Okuma Süreci Hastalık Araştırmalarına Yeni Bir Pencere Açıyor
 Kolorektal kanserde ameliyat sonrası riskleri yaş ve tümör evresi birlikte şekillendiriyor
Kolorektal kanserde ameliyat sonrası riskleri yaş ve tümör evresi birlikte şekillendiriyor